SEOUL, August 10 (AJP) - KAIST scientists have unveiled an artificial intelligence model that can automatically design potential drug molecules precisely tailored to disease-causing proteins, even when no prior information about suitable molecules is available. The breakthrough could speed up drug discovery and cut costs, especially for hard-to-treat cancers.
Traditionally, drug development starts by identifying a target protein, such as a cancer cell receptor, then screening vast libraries of molecules to find one that binds effectively to block its harmful activity. This process is time-consuming, expensive, and has a low success rate. The new KAIST system, called "BInD" (Bond and Interaction-Generating Diffusion model), skips the trial-and-error search by generating both the molecular structure and the way it interacts with the target protein in a single step.
The team, led by Kim Woo-youn of KAIST's Department of Chemistry, designed BInD to create molecules that satisfy multiple drug design requirements at once, such as stability, physical properties, and structural realism. Most existing AI models generate molecules separately and then evaluate how they might bind to proteins. BInD instead considers the protein–molecule binding process from the start, boosting the chances of producing effective and stable candidates.
The AI uses a "diffusion model", the same approach that underpinned the 2024 Nobel Prize in Chemistry for the AlphaFold3 system for protein–drug structure prediction. Unlike AlphaFold3, which predicts atomic positions directly, BInD integrates knowledge-based chemical guidelines, such as bond lengths and distances between atoms, to ensure its generated molecules follow real-world chemical laws.
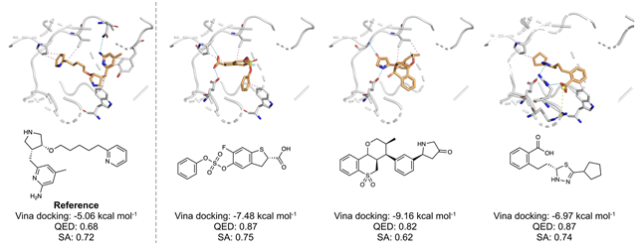
The researchers also developed an optimization strategy that reuses strong binding patterns found in earlier AI-generated results, enabling the system to improve output without additional training. Using this method, the team successfully designed molecules that selectively target mutant forms of EGFR, a protein linked to certain cancers.
This work builds on the group's previous AI models, which required pre-defined information on how molecules should bind to proteins. BInD removes that dependency, learning the key binding factors on its own.
"This AI can understand and learn the essential elements for binding to a target protein, enabling it to design optimal drug candidates without prior molecular information," said Kim. "It has the potential to transform drug discovery by making it faster, more precise, and more reliable."
The findings, co-authored by doctoral candidates Lee Joong-won and Jeong Won-ho as first authors, were published on Jul. 11 in the journal Advanced Science under the title "BInD: Bond and Interaction-Generating Diffusion Model for Multi-Objective Structure-Based Drug Design."

Copyright ⓒ Aju Press All rights reserved.